Introduction
CRISPR imaging is a CRISPR-based technology that can track the genomic elements in real-time and provide their spatial and temporal information. In CRISPR imaging systems, single guide RNAs (sgRNA) direct the imaging probes, such as fluorescent proteins, to the genomic loci of interest. These systems necessitate clustered binding sites within the target areas and low off-target effects outside these target regions.
CRIBAR is a fast and flexible sgRNA design tool tailored for CRISPR imaging experiments. It crafts an optimal set of sgRNAs to reduce the cost and boost the efficiency of the CRISPR-based imaging experiment. CRIBAR formulates two optimization scenarios.
- Formulation 1: Given the minimum on-target activity score, output the minimal set of sgRNAs.
- Formulation 2: Given the maximum number of sgRNAs, output a set of sgRNAs with the maximum on-target activity score.
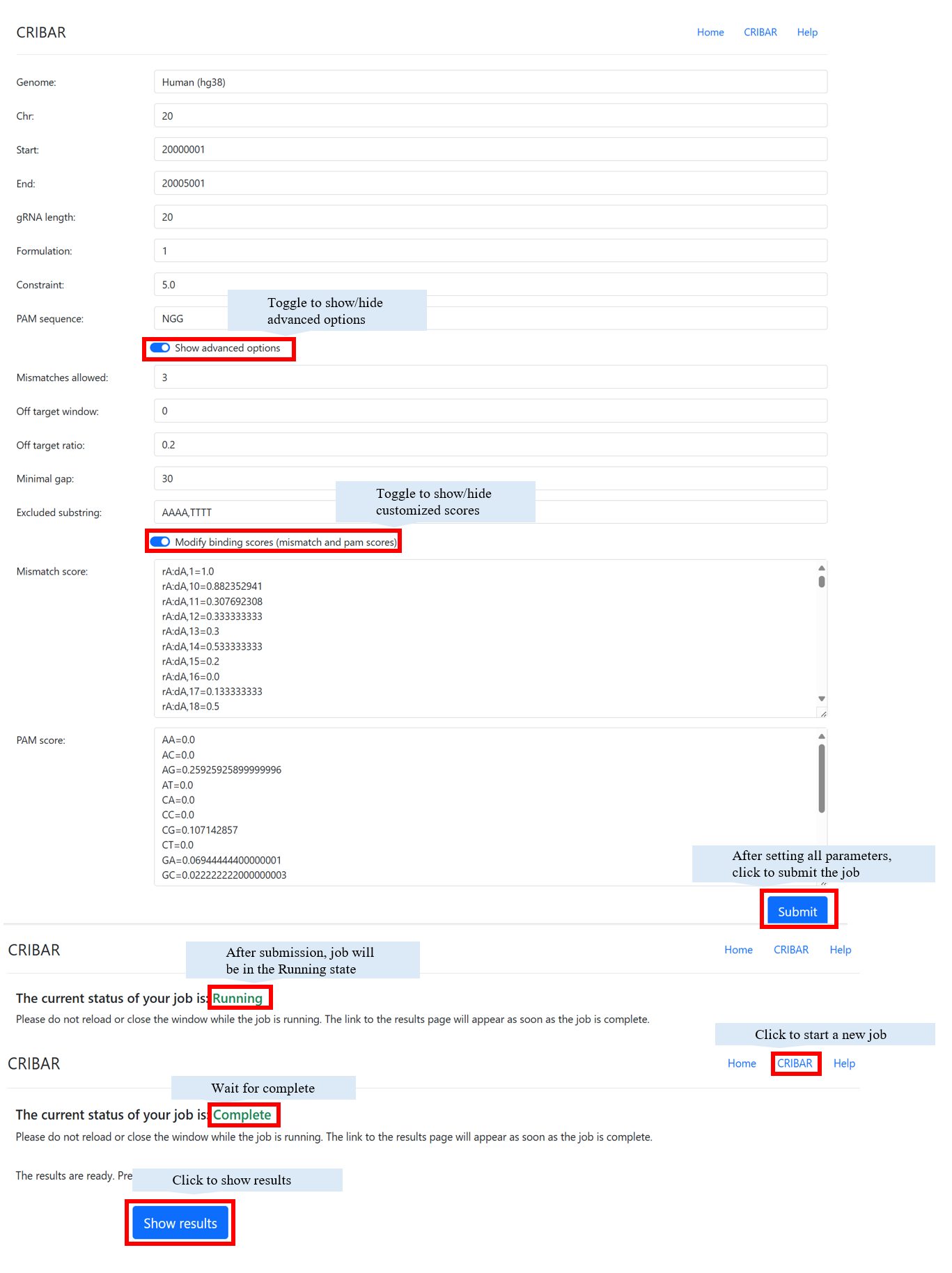
Parameters
| genome: | The reference genome used in CRISPR imaging experiment design. |
| chr: | The chromosome of the target region. |
| start: | The starting position of target region. |
| end: | The ending position of target region. |
| len: | The length of gRNA (not include pam). |
| formulation: | 1 - Given the minimum on-target activity score, output the minimal set of sgRNAs. 2 - Given the maximum number of sgRNAs, output a set of sgRNAs with the maximum on-target activity score. |
| constraint: | When formulation=1, constrain is the minimum on-target activity score. When formulation=2, constrain is the maximum gRNA number. |
| pam: | Protospacer adjacent motif. Default is NGG. |
| mismatch: | The number of mismatches allowed in each gRNA. |
| off_target_window: | The window size that CRIBAR uses to check the off-targets. Default value is the same length of the target region. |
| off_target_ratio: | The threshold of (off-target binding site density) / (on-target binding site density). |
| min_gap: | The minimum distance between two binding sites. |
| excluded_substring: | Excluded substrings separated by a comma. Example: AAAA,TTTT. |
| mismatch and pam scores: | If you want to use your own scores instead of the CFD scores, you can modify the files mismatch_score and pam_score in the score folder. CRIBAR will read the scores from these two files. |
Source code
CRIBAR tool is developed using Python 3 and the latest source code will be found in the following GitHub repository: github.com/ucfcbb/CRIBAR.
Cite this work
If you use CRIBAR in your work, please cite: Xiaoli Chen, Md Mahfuzur Rahaman, Ardalan Naseri and Shaojie Zhang. CRIBAR: a fast and flexible sgRNA design tool for CRISPR imaging.
For bug reports or comments please contact: xiaolichen.cs@gmail.com or mahfuz@ucf.edu.